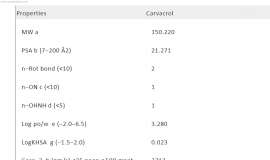
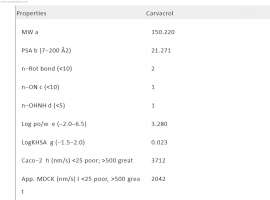
SRC signaling mediates acquired resistance in ALK-positive NSCLC
Multiple SRC family kinase inhibitors were consistently ef-fective across several patient-derived ALK-positive resistant NSCLC models (Fig. 2). In particular, AZD0530 (saracatinib) was a hit in 6 of 9 patient-derived ALK lines tested (Fig. 2A). Models in which AZD0530 was a screen hit had unremarka-ble sensitivity to single agent AZD0530 indicating that, as in other cases, these cell lines have not switched to an entirely different dependency. On the other hand these resistant ALK-positive cell lines were highly sensitive to AZD0530 in the presence of ALK inhibitors (Fig. 4A). Drug synergism between AZD0530 and ALK inhibitors was also observed (average of 20% less viability than expected across all con-centrations for five models retested in triplicate with maxi-mum differences ranging from 18 to 45% over Bliss (table S8). Two other drugs (dasatinib and KIN001-113) that po-tently inhibit SFKs (23, 24) were often hits in models in which AZD0530 was a hit (Fig. 2B and fig. S5). However, due to the more favorable specificity profile of AZD0530 (25), we used this drug in our subsequent studies. Each model in which AZD0530 was a hit (as indicated by arrows in Fig. 4A) was significantly sensitized to ALK inhibition by AZD0530 (Fig. 4B). Notably, other ALK driven models also demonstrated shifts in sensitivity with AZD0530 pointing to the possibility of broad involvement of SRC kinases in ALK inhibitor response. Interestingly, AZD0530 was not a hit in any of the mutant EGFR or HER2 amplified cancers and in only 1 of 9 MET amplified cancers (fig. S5).
We next aimed to determine the relevant target of AZD0530. Overexpression of the kinase-dead SRC K295R (26), as well as knockdown of SRC alone with either of two shRNAs effectively recapitulated the effect of AZD0530, demonstrating that among AZD0530 targets, including mul-tiple SFKs, SRC inhibition is sufficient to resensitize cells to ALK inhibition (Fig. 4C). We observed that multiple ALK driven models were sensitive to both SRC and EGFR inhibi-tors when combined to an ALK inhibitor. However, the ac-tivity of AZD0530 does not appear to be driven by EGFR inhibition directly or indirectly since AZD0530 did not in-hibit EGFR activation in the ALK-positive MGH025-1A cells, which were sensitized by AZD0530 (fig. S10A). Furthermore, some cell lines, such as MGH010-1A, were sensitized by AZD0530, but not EGFR inhibitors (Fig. 2A and fig. S10B). We next examined the effect of combined ALK and SRC in-hibition on three resistant ALK-positive models derived from patient biopsies: MGH010-1A and MGH025-A (re-sistant to crizotinib, no ALK resistance mutations) and MGH049-1A [resistant to ceritinib, no ALK resistance muta-tions (27)]. In all three models, cells grew at 6 days when treated with either drug as single agent, but combination treatment resulted in loss of cell viability compared to pre-treatment cell number (Fig. 4D) and robust apoptotic cell death (S11A). Consistent with these results, the ALK TKI failed to fully inhibit downstream signaling (AKT, MAPK or S6K) except in the presence of AZD0530 in each of these resistance models (Fig. 5A and fig. S11B).
In each of the patient-derived ALK models in which AZD0530 was effective (including MGH034-2A, which nar-rowly failed to meet our threshold for hit call for AZD0530), ALK inhibition resulted in robust up-regulation of SRC ac-tivity as measured by the phosphorylation of the SRC sub-strate Paxillin (Fig. 5B). Thus, ALK inhibition may lead to up-regulation of SRC signaling, perhaps via release of a neg-ative regulatory signal normally coordinating ALK and SRC activities. In contrast, we did not consistently observe an increase in SRC activity as measured by p-Paxillin in EGFR mutant cancers following EGFR inhibitor treatment (fig. S11C), consistent with the absence of efficacy noted with AZD0530 in EGFR mutant cancer. Furthermore, in the ALK driven models, SRC signaling was also up-regulated by inhi-bition of signaling pathways downstream of ALK. Interest-ingly, although the downstream pathways regulated by ALK in individual models vary, the pathways regulated by ALK tended to be the one suppressing SRC signaling. For exam-ple, when ALK inhibition primarily impacted PI3K signaling but not MEK activity, PI3K inhibition up-regulated SRC sig-naling (fig. S12A). Moreover, when ALK inhibition sup-pressed both MAPK and PI3K signaling, SRC signaling was robustly up-regulated by either PI3K or MAPK signaling (fig. S12B). Overall these results are compatible with a mod-el in which ALK activity suppresses SRC activity broadly in the setting of ALK-driven cancers.
To further characterize the effect of ALK inhibition on these models, we performed gene expression analysis on each of the ALK-positive patient-derived models in the pres-ence or absence of an ALK inhibitor for 24 hours. The gene ontologies most enriched within genes whose expression was induced by ALK inhibition were extracellular matrix and basal membrane (Benjamini-Hochberg corrected p val-ues 1.75E-04 and 2.31E-04) (Fig. 5C and databases S6 to S8). As SRC signaling is known to be a focal point of integrin-mediated signaling and the transduction of extracellular signals, these results further support the finding that SRC activity is increased upon inhibition of ALK signaling in ALK-positive lung cancers.
Finally, we tested the efficacy of the combination of ALK TKIs and AZD0530 in vivo using mouse xenograft models. In MGH025-1A (derived from an ALK-positive patient who had become resistant to crizotinib), treatment with single-agent crizotinib resulted in tumor progression after 34 days. However, combining AZD0530 and crizotinib resulted in a sustained, profound response for over 60 days (Fig. 5D). Notably, when AZD0530 was added to the treatment of the xenografts that had progressed on crizotinib, the tumors regressed (fig. S13A). To test the specificity of AZD0530 for resistant models that demonstrated synergy in the screen, we tested it in the HCC827 GR6 line, which harbors a MET bypass track and was not a hit for AZD0530. In this model the combination of AZD0530 with gefitinib was ineffective in comparison to gefitinib plus crizotinib (which is a potent MET inhibitor) (fig. S13B). Thus, the effect of AZD0530 ap-pears particular to the models in which combination effica-cy was found in the screen.
Analysis of the discovered mutations identified by the 1,000-gene NGS panel in the ALK-positive models failed to identify mutations in SRC family kinases and other known regulators of SRC activity (table S7). Thus, the pharmacolog-ic approach identified a drug combination that would not have readily been predicted by genomic analyses alone.
 Science-2014-Crystal-science.1254721.pdf
(1.68 MB, 下载次数: 37)
Science-2014-Crystal-science.1254721.pdf
(1.68 MB, 下载次数: 37)
|




 显身卡
显身卡